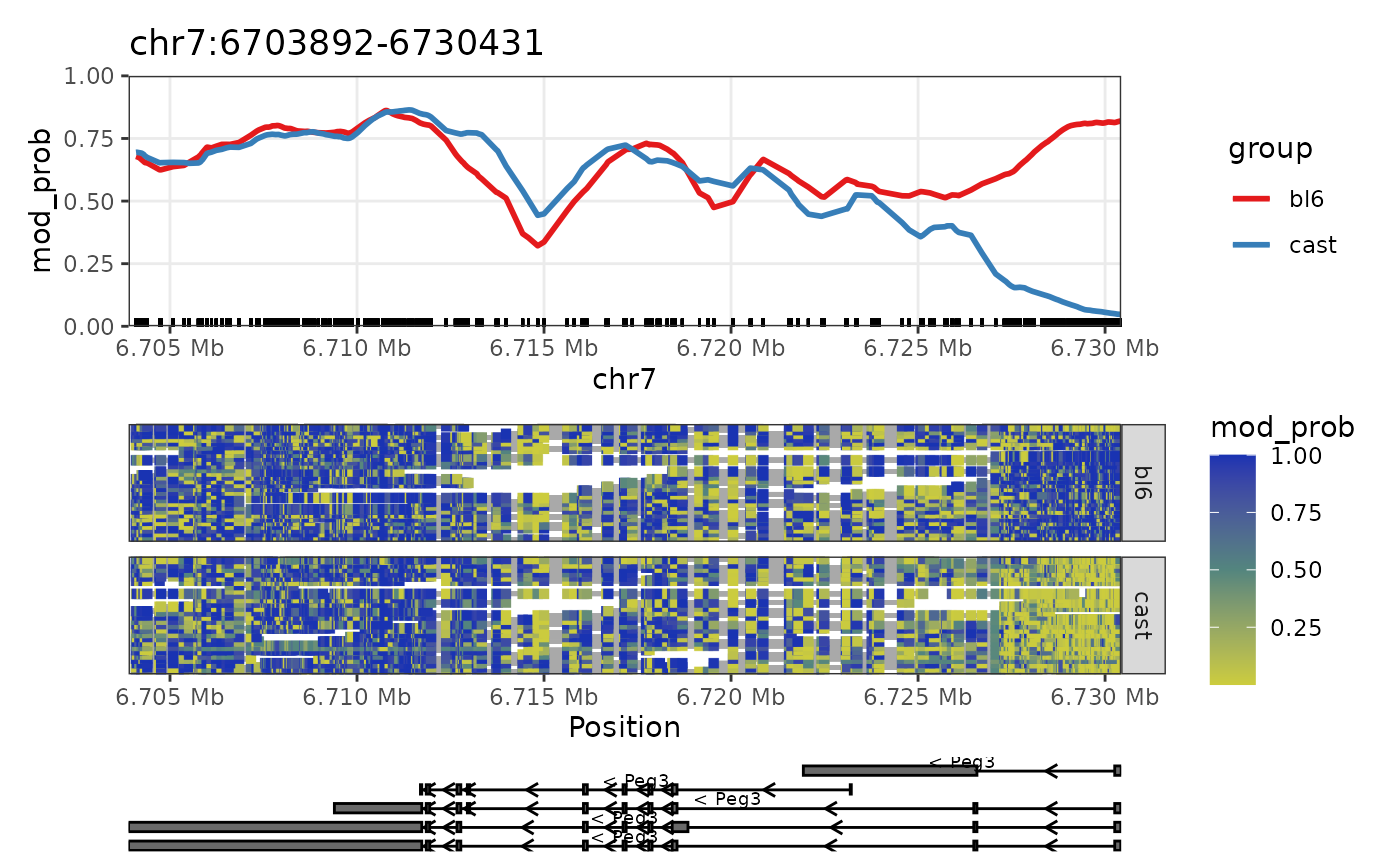
Plot GRanges
Usage
plot_grange(
x,
grange,
anno_regions = NULL,
binary_threshold = NULL,
avg_method = c("mean", "median"),
spaghetti = FALSE,
heatmap = TRUE,
heatmap_subsample = 50,
gene_anno = TRUE,
smoothing_window = 2000,
window_prop = 0,
palette = ggplot2::scale_colour_brewer(palette = "Set1"),
line_size = 1,
span = NULL
)Arguments
- x
the NanoMethResult object.
- grange
the GRanges object with one entry.
- anno_regions
the data.frame of regions to be annotated.
- binary_threshold
the modification probability such that calls with modification probability above the threshold are set to 1 and probabilities equal to or below the threshold are set to 0.
- avg_method
the average method for pre-smoothing at each genomic position. Data is pre-smoothed at each genomic position before the smoothed aggregate line is generated for performance reasons. The default is "mean" which corresponds to the average methylation fraction. The alternative "median" option is closer to an average within the more common methylation state.
- spaghetti
whether or not individual reads should be shown.
- heatmap
whether or not read-methylation heatmap should be shown.
- heatmap_subsample
how many packed rows of reads to subsample to.
- gene_anno
whether to show gene annotation.
- smoothing_window
the window size for smoothing the trend line.
- window_prop
the size of flanking region to plot. Can be a vector of two values for left and right window size. Values indicate proportion of gene length.
- palette
the ggplot colour palette used for groups.
- line_size
the size of the lines.
- span
DEPRECATED, use smoothing_window instead. Will be removed in next version.
Examples
nmr <- load_example_nanomethresult()
#> Successfully matched 6 samples between data and annotation.
plot_grange(nmr, GenomicRanges::GRanges("chr7:6703892-6730431"))