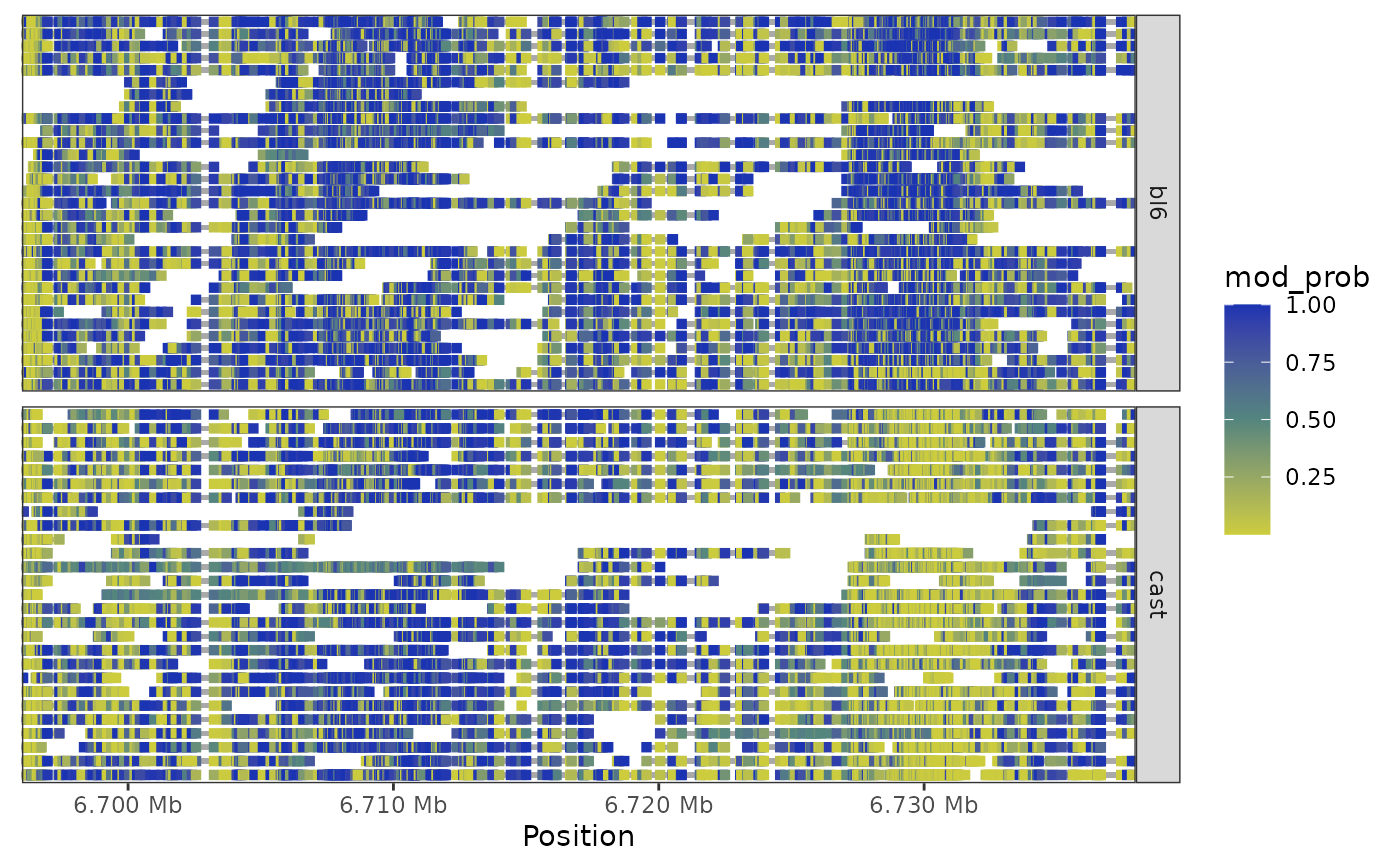
Plot the methylation heatmap of a gene symbol specified within the exon(x) slot.
Usage
plot_gene_heatmap(x, gene, ...)
# S4 method for class 'NanoMethResult,character'
plot_gene_heatmap(
x,
gene,
window_prop = 0.3,
pos_style = c("to_scale", "compact"),
subsample = 50
)
# S4 method for class 'ModBamResult,character'
plot_gene_heatmap(
x,
gene,
window_prop = 0.3,
pos_style = c("to_scale", "compact"),
subsample = 50
)Arguments
- x
the NanoMethResult or ModBamResult object.
- gene
the gene symbol for the gene to plot.
- ...
additional arguments.
- window_prop
the size of flanking region to plot. Can be a vector of two values for left and right window size. Values indicate proportion of gene length.
- pos_style
the style for plotting the base positions along the x-axis. Defaults to "to_scale", plotting (potentially) overlapping squares along the genomic position to scale. The "compact" options plots only the positions with measured modification.
- subsample
the number of read of packed read rows to subsample to.
Details
This function creates a heatmap visualisation of methylation data for a specific gene. Each row in the heatmap represents one or more packed reads, where colored segments indicate methylation probability at each genomic position.
Examples
nmr <- load_example_nanomethresult()
#> Successfully matched 6 samples between data and annotation.
plot_gene_heatmap(nmr, "Peg3")