Plot gene aggregate plot
Usage
plot_agg_genes(
x,
genes = NULL,
binary_threshold = 0.5,
group_col = NULL,
flank = 2000,
stranded = TRUE,
span = 0.05,
palette = ggplot2::scale_colour_brewer(palette = "Set1")
)Arguments
- x
the NanoMethResult or ModBamResult object.
- genes
a character vector of gene symbols to include in aggregate plot. If NULL (default), all genes in exons(x) are used.
- binary_threshold
the modification probability such that calls with modification probability above the threshold are considered methylated, and those with probability equal or below are considered unmethylated.
- group_col
the column name to group aggregated trends by. This column can be found in either the regions table or samples(x). When NULL (default), all data is aggregated together. Common values include "sample" to show individual samples or "group" to show sample groups.
- flank
the number of flanking bases to add to each side of each region.
- stranded
if TRUE, negative strand features will have their coordinates flipped to reflect biological features like transcription start sites (e.g., for genes, coordinates run from TSS to TES regardless of strand).
- span
the span parameter for loess smoothing of the trend lines.
- palette
the ggplot colour palette used for groups.
Details
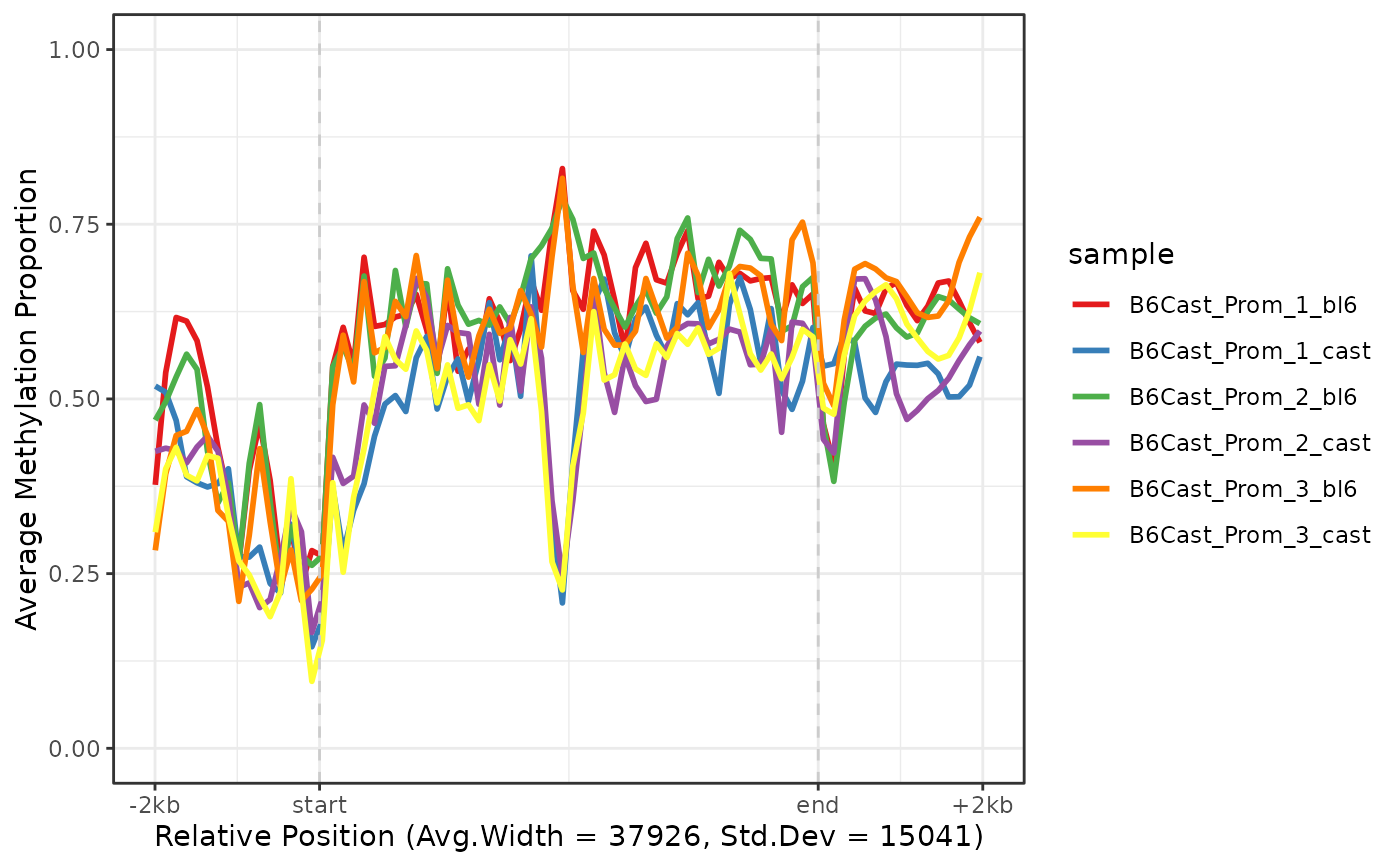
This function creates an aggregate methylation profile across multiple genes by scaling all genes to the same relative coordinates (0 to 1) and averaging methylation levels at each relative position. Genes are optionally extended by flanking regions specified by the flank parameter. The resulting plot shows smoothed trends of average methylation probability from gene start to gene end, with optional flanking regions.
Examples
nmr <- load_example_nanomethresult()
#> Successfully matched 6 samples between data and annotation.
plot_agg_genes(nmr)
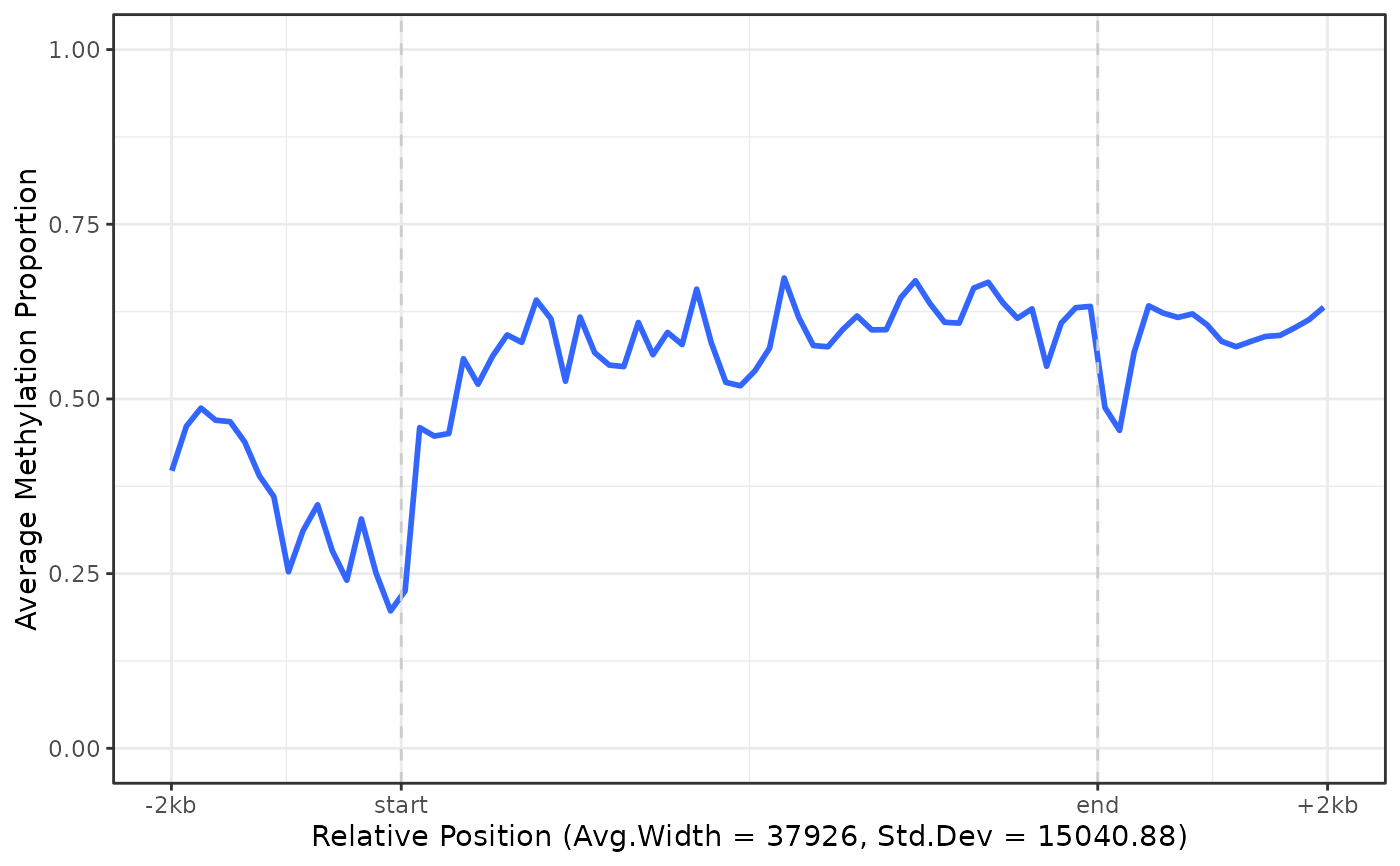 # Plot specific genes only
plot_agg_genes(nmr, genes = c("Peg3", "Impact"))
# Plot specific genes only
plot_agg_genes(nmr, genes = c("Peg3", "Impact"))
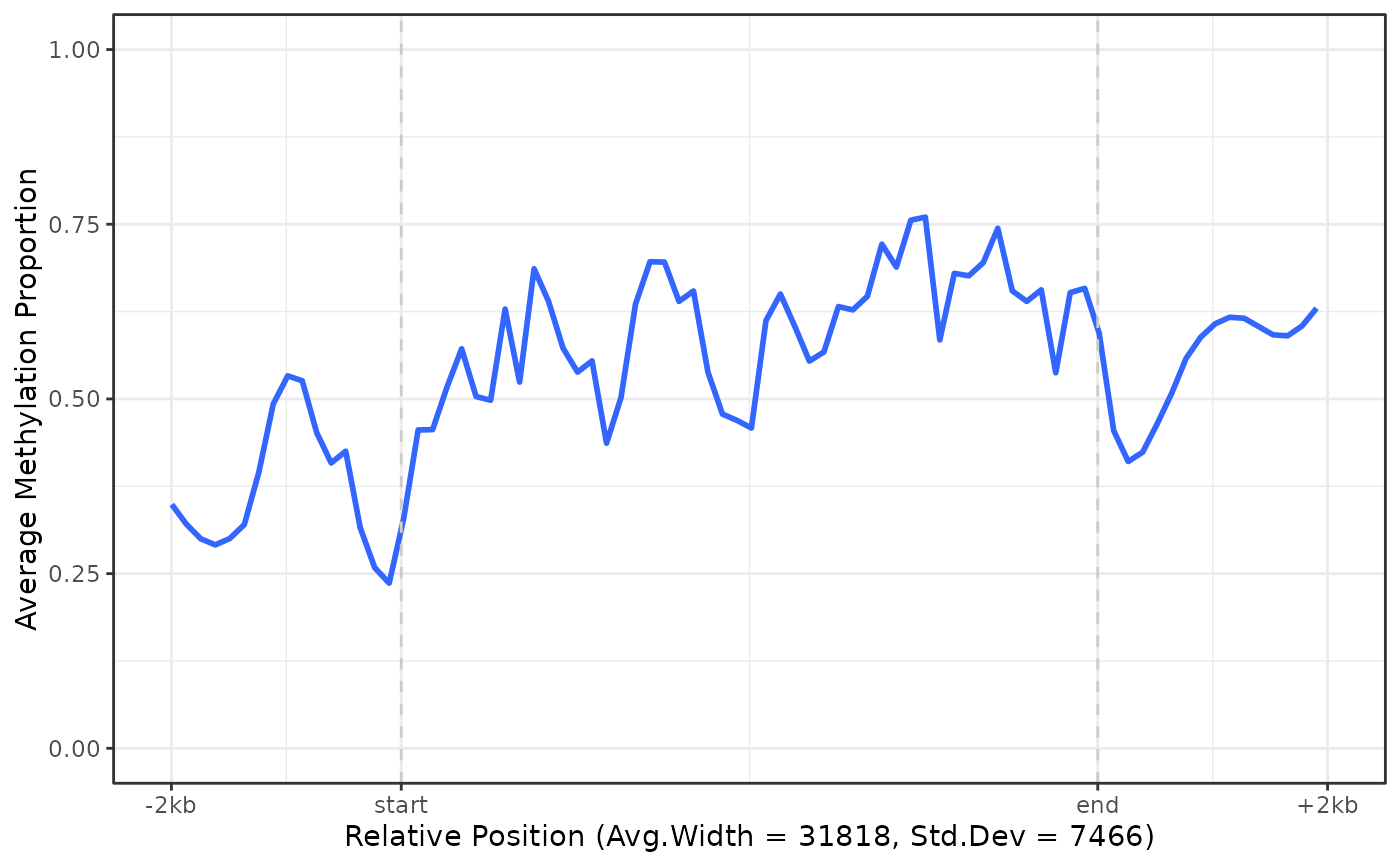 # Group by sample
plot_agg_genes(nmr, group_col = "sample")
# Group by sample
plot_agg_genes(nmr, group_col = "sample")