Chapter 2 Session 2
In the previous session we learned about:
- The RStudio environment
- Mathematical and logical operations
- Using functions
- Variables
- Atomic and collection data types
- Input and output
In this session we will learn about:
- Missing values
- Subsetting data structures
- How to merge two dataframes
2.1 Missing values
Missing values are fairly common in data, we will look at how to deal with missing values in R. First, let’s read in some data. Recall we use the function read.delim() and tell R not to read strings as factors using stringsAsFactors = FALSE.
We are using the file “Ses2_genes.tsv” today.
genes <- read.delim("data/Ses2_genes.tsv", stringsAsFactors = FALSE)
dim(genes) # returns number of rows and columns## [1] 200 5## EntrezID SYMBOL TXCHROM GeneLength Count
## 1 11480 Acvr2a chr2 5681 979
## 2 11634 Aire chr10 1929 15
## 3 12156 missing chr2 2515 932
## 4 12290 Cacna1e chr1 12697 0
## 5 12449 Ccnf chr17 3099 214
## 6 12563 Cdh6 chr15 2632 2You can see in the environment tab that this data frame has 200 rows (observations) and 5 columns (variables). You can also click on the genes entry in the Environment tab (red arrow in Figure 2.1) to display the data in a new window (left):
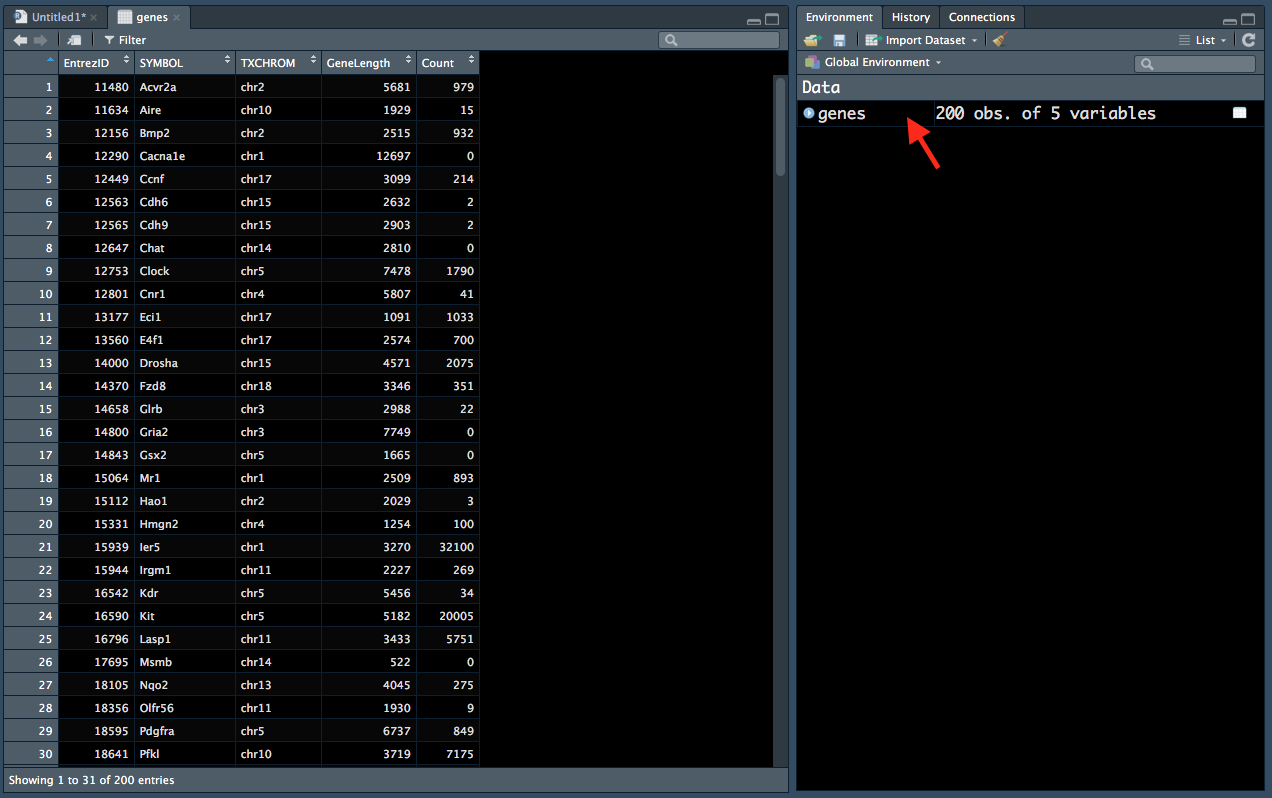
Figure 2.1: Viewing data from the ‘Environment’ tab.
Scrolling down the window, you can see that there are several NA entries. NA denotes a missing value in R.
NA‘s have some interesting behaviour - they are ’contagious’. For example, if we wanted to take the mean of a vector numbers, which includes a NA, the answer is NA. If we think about it, it makes sense that the mean of two numbers and a ‘missing’ number, that could be anything, is NA.
## [1] NALuckily many mathematical functions have a na.rm option, where you can tell it to remove NA values:
## [1] 2.5Finding out if something is NA is also a bit different, due to the nature of the missing value. So we must use the is.na() function rather than a direct comparison.
## [1] NA## [1] TRUE## [1] FALSE FALSE TRUEOne last thing to note is that in our data, the symbol of the third row is “missing”, and the 30th row is “Missing”. These are other representations of missing values found in our data that is not detect as such by R. We can change the value to NA in our original data, but generally we don’t want to edit our raw data. Instead we can tell read_delim() what values should be interpreted as NA via the na.strings argument.
2.2 Subsetting vectors
Subsetting involves selecting a portion of a data structure and uses square brackets [ ]. There are two main ways to subset a data structure: numerical or logical indices.
2.2.1 Subsetting with numeric indicies
Let’s take a look at subsetting vectors first. We can use $ to obtain just one column from the genes data frame. The output will be a vector.
## [1] 979 15 932 0 214 2 2 0 1790 41 1033 700
## [13] 2075 351 22 0 0 893 3 100 32100 269 34 20005
## [25] 5751 0 275 9 849 7175 768 358 234 5065 2096 1994
## [37] 757 0 2310 0 1 0 3091 810 30 816 817 1
## [49] 580 33 0 941 1445 522 2 2346 572 431 5797 2
## [61] 70 1236 966 235 2 3009 431 452 2602 3481 99 11857
## [73] 35 1952 0 706 1317 1130 92 0 871 730 3892 3772
## [85] 20 7 0 756 1 210 101 17 21 537 1240 930
## [97] 1 1 0 221 1599 0 67 1 0 315 0 0
## [109] 739 0 0 976 1 715 18636 289 1396 0 101 285
## [121] 1665 47 2 4483 2 363 169 135 0 2 0 11
## [133] 4088 1082 92 0 3888 17 0 0 0 0 0 0
## [145] 0 0 0 0 2442 30 1610 0 0 0 16 1
## [157] 2 0 47 0 1118 0 1 0 2 3 22 0
## [169] 0 0 0 0 0 0 431 0 0 16 0 1
## [181] 0 4 0 0 1 0 0 0 0 301 0 0
## [193] 0 0 0 0 0 0 0 59We will assign this to a variable called counts. counts is now a variable that refers to a vector containing 200 integers.
## [1] 979 15 932 0 214 2 2 0 1790 41 1033 700
## [13] 2075 351 22 0 0 893 3 100 32100 269 34 20005
## [25] 5751 0 275 9 849 7175 768 358 234 5065 2096 1994
## [37] 757 0 2310 0 1 0 3091 810 30 816 817 1
## [49] 580 33 0 941 1445 522 2 2346 572 431 5797 2
## [61] 70 1236 966 235 2 3009 431 452 2602 3481 99 11857
## [73] 35 1952 0 706 1317 1130 92 0 871 730 3892 3772
## [85] 20 7 0 756 1 210 101 17 21 537 1240 930
## [97] 1 1 0 221 1599 0 67 1 0 315 0 0
## [109] 739 0 0 976 1 715 18636 289 1396 0 101 285
## [121] 1665 47 2 4483 2 363 169 135 0 2 0 11
## [133] 4088 1082 92 0 3888 17 0 0 0 0 0 0
## [145] 0 0 0 0 2442 30 1610 0 0 0 16 1
## [157] 2 0 47 0 1118 0 1 0 2 3 22 0
## [169] 0 0 0 0 0 0 431 0 0 16 0 1
## [181] 0 4 0 0 1 0 0 0 0 301 0 0
## [193] 0 0 0 0 0 0 0 59This will give you the 3rd element of the vector counts:
## [1] 932It is also possible to give a vector of indices, this returns a vector of the same length as the indices given containing the elements corresponding to each index.
## [1] 932 0 214 2 2 0 1790 41The indices do not need to be sequential, nor do they need to be unique or in ascending order.
## [1] 932 214 221## [1] 932 932 214## [1] 221 932 214Note that we have used c() within the square brackets. This is because R expects ONE ‘object’ within the square brackets. Thus, if you want to extract several indices, you must give it ONE vector containing the indices of the elements. A vector (e.g. c(3,5,10)) is considered one ‘object’ but the numbers 3,5,10 are considered three different ‘objects’.
Indeed, 3:10, which we used earlier, is actually a vector of the numbers 3 to 10:
## [1] 3 4 5 6 7 8 9 10We can also use the - sign to index all the elements EXCEPT the elements 10 through to 200:
## [1] 979 15 932 0 214 2 2 0 1790Note that subsetting past the end of a vector returns NA.
## [1] NA## [1] "a" "c" NA2.2.2 Subsetting with logical vectors
The alternative way to subset is using a logical vector of the same length as the vector being indexed. Each element of the logical vector denotes whether or not that element of the original vector will be returned in the result.
## [1] "a" "b" "d"You will rarely write out these logicals by hand, instead they usually come from a comparisons, for example if we wanted to take only the non-zero values of counts
## [1] 979 15 932 214 2 2 1790 41 1033 700 2075 351
## [13] 22 893 3 100 32100 269 34 20005 5751 275 9 849
## [25] 7175 768 358 234 5065 2096 1994 757 2310 1 3091 810
## [37] 30 816 817 1 580 33 941 1445 522 2 2346 572
## [49] 431 5797 2 70 1236 966 235 2 3009 431 452 2602
## [61] 3481 99 11857 35 1952 706 1317 1130 92 871 730 3892
## [73] 3772 20 7 756 1 210 101 17 21 537 1240 930
## [85] 1 1 221 1599 67 1 315 739 976 1 715 18636
## [97] 289 1396 101 285 1665 47 2 4483 2 363 169 135
## [109] 2 11 4088 1082 92 3888 17 2442 30 1610 16 1
## [121] 2 47 1118 1 2 3 22 431 16 1 4 1
## [133] 301 59## [1] 979 15 932 214 2 2 1790 41 1033 700 2075 351
## [13] 22 893 3 100 32100 269 34 20005 5751 275 9 849
## [25] 7175 768 358 234 5065 2096 1994 757 2310 1 3091 810
## [37] 30 816 817 1 580 33 941 1445 522 2 2346 572
## [49] 431 5797 2 70 1236 966 235 2 3009 431 452 2602
## [61] 3481 99 11857 35 1952 706 1317 1130 92 871 730 3892
## [73] 3772 20 7 756 1 210 101 17 21 537 1240 930
## [85] 1 1 221 1599 67 1 315 739 976 1 715 18636
## [97] 289 1396 101 285 1665 47 2 4483 2 363 169 135
## [109] 2 11 4088 1082 92 3888 17 2442 30 1610 16 1
## [121] 2 47 1118 1 2 3 22 431 16 1 4 1
## [133] 301 59Indices are like shopping lists, each element of the index tell us what we need from the original vector. In numerical indices it tells us the position in the vector to take values from, and in logical indices it tells us whether each element should be included in the result.
2.2.3 Useful summaries of logical vectors
Two relevant function that are useful to know is the which() and table(). which() takes a logical vector and tells you the indices of the TRUE elements. table() takes any vector and gives you counts of the unique elements, for logical vectors this counts the TRUE and FALSE.
## [1] 1 4##
## FALSE TRUE
## 2 2Challenge 2.1
- Predict the return values of the following expressions.
# i.
x <- c("a", "b", "c")
x[c(2, 1, 1, 3)]
# ii.
c(1, 2, 3, NA) + 1
# iii.
x <- c("a", "b", "c")
x[c(TRUE, FALSE, TRUE, TRUE)]- Take the following vector and create two subsets of it that are <=5 and >5. How many elements are in each subset?
vec <- c(5.16, 7.09, 8.08, 7.6, 3.04, 9.35, 4.68, 8.29, 8.26, 7.38,
5.87, 7.43, 4.52, 6.09, 8.52, 6.03, 4.05, 6.11, 0.9, 2.79, 1.54,
4.57, 3.51, 1.95, 5.72, 6.04, 4.29, 3.47, 1.76, 5.25, 9.91, 2.73,
7.42, 4.85, 6.41, 5.2, 5.8, 3.22, 8.18, 9.96
)- Using numeric indices, create the word
c("h", "e", "l", "l", "o")from the following vector. i.e. something of the formletter_vec[c(...)].
2.2.4 Overwriting subset values
You can assign to a subset of a vector to change the values of that subset within a vector.
## [1] 1 2 10 4 10This also works with logical vectors in the similar way.
## [1] 1 2 10 4 102.3 Subsetting data structures
Subsetting a 2 dimensional data structure (e.g. a data frame or a matrix) is similar to subsetting a vector, except you now must specify which rows AND which columns you want. The general syntax for the genes data frame looks like this:
genes[(rows you want) , (columns you want)]Within the square brackets, you will specify which rows you want, followed by a comma, then what columns you want.
2.3.1 Subsetting data structures with numerics
As with vectors, you can use numeric vectors to select the rows and columns you want.
## SYMBOL GeneLength
## 5 Ccnf 3099
## 6 Cdh6 2632
## 7 Cdh9 2903
## 8 Chat 2810
## 9 Clock 74782.3.2 Subsettting data frames using characters
Recall that we could select individual columns of the data frames using the $ operator along with the column name. We can also select multiple columns using a vector of column names. Note that column names are enclosed in quotes signifying that they are of the ‘character’ data type.
## SYMBOL GeneLength
## 5 Ccnf 3099
## 6 Cdh6 2632
## 7 Cdh9 2903
## 8 Chat 2810
## 9 Clock 7478If you leave the left side of comma empty, R will give you ALL the rows. If you leave the right side of the comma empty, R will give you ALL the columns.
This will give you only two columns of the data frame.
This will give you the 2nd row and all the columns.
## EntrezID SYMBOL TXCHROM GeneLength Count
## 2 11634 Aire chr10 1929 152.3.3 Removing NA rows using complete.cases()
Because of the care required when working with missing values, it is sometimes an option to simply remove all entries that are not complete. This can be done using the complete.cases() function. This function returns a logical vector indicating whether each row of a data frame contains no missing values.
# returns logical of same length as number of rows in genes
complete.cases(genes)
# can be used to filter rows down to only complete rows
complete_genes <- genes[complete.cases(genes), ]
complete_genesChallenge 2.2
- Take the following counts vector and overwrite all values below 10 with the value 0.
- Take the following character vector and using a subset reasisgnment, create the word
c("g", "e", "n", "e", "t", "i", "c", "s").
- Take the
genesdata frame and select the symbol, chromosome and count columns of only complete cases that are not missing gene symbols.
2.3.4 Rearranging using the order() function
We have seen before that numeric indices can be used to change the order of the output relative to the original input. A common application of this is to arrange a table by the values in some column using the order() function.
The order function returns a vector of numerics of the same length with values such that the the vector would be sorted.
# order returns a vector of numerics that can be use to index
order(genes$TXCHROM)
# using this to index will result in a sorted vector
genes$TXCHROM[order(genes$TXCHROM)]
# it can also be applied to data frames to sort by a column
genes[order(genes$TXCHROM), ]
# multiple arguments can be given to order to sort by multiple columns
genes[order(genes$TXCHROM, genes$SYMBOL), ]2.3.5 Subsetting data structures with logicals
One very useful operation is to subset a whole table based on the values of a particular column.
As an example, we subset our genes table to only the genes with a count of at least 100.
# subset all the genes with 0 counts and every column
genes[genes$Count == 0, ]
# subset all the genes with >0 counts and every column
genes[genes$Count > 0, ]This allows tables to be filtered based on a specific column.
2.3.6 %in%, & and |
The %in% operator tests whether elements of the first vector is contained within the second vector. It will return TRUE where the elements of the first vector is found in the second.
# %in% operator tests membership
c("a", "b", "c", "d") %in% c("b", "d")
# result of in can be used as a filtering for multiple matches
genes$TXCHROM %in% c("chr1", "chr2")
genes[genes$TXCHROM %in% c("chr1", "chr2"), ]The & (AND) operator takes two logical vectors and performs an AND operation
on them, this means that along the positions of the two vectors, TRUE is
returned only if it is TRUE in those positions of both vectors.
Rather than thinking about the logcail AND operation, it’s easier to just read the expression literally. For example if we wanted to select genes from chromosome 1 and with counts greater than 500, we use the following row index.
The | (OR) is a complementary operation that takes two logical and performs an
OR operation, this returns TRUE if one or both values are TRUE. Once again we
can simply read the expression literally. If we wanted genes that are from
chromosome 1 or have counts greater than 500 we can use the following row index.
2.4 Merge
Two dataframes can be combined with the merge() function.
For example, let’s say we have two dataframes (dataframe1 and dataframe2), each containing different information about 3 genes:
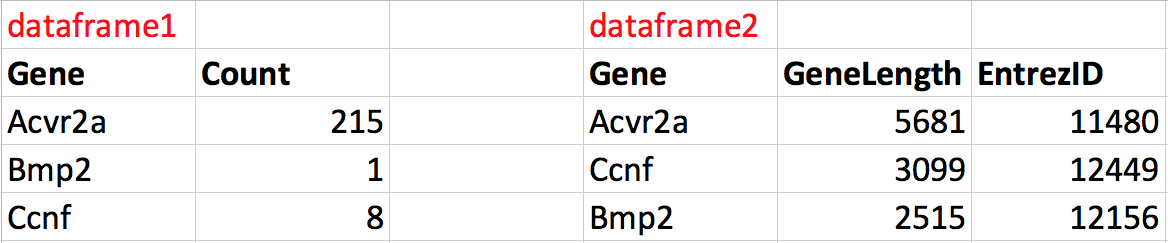
Figure 2.2: The two dataframes to merge.
We can merge these dataframes together into one data frame that contains all the information about genes. Notice that the order of the genes is not the same in the two dataframes. During the merge we want R to match each row according to the Gene column in each data frame such that the correct information is added to the correct row. The result would have 4 columns, 3 rows and the correct information along each row.
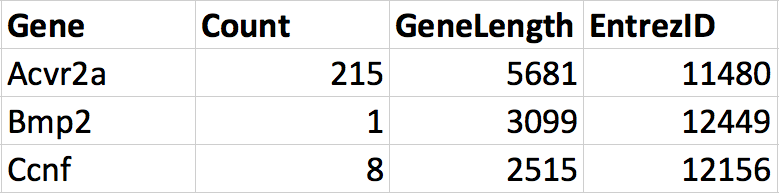
Figure 2.3: The two dataframes merged.
Let’s practice merging on the files “Ses2_genes.tsv” and “Ses2_geneNames.tsv”. “Ses2_genes.tsv” contains gene EntrezIDs, gene symbol, gene chromosome, gene length and their count value. “Ses2_geneNames.tsv” contains gene names and their corresponding EntrezIDs.
First we will read in both files:
genes <- read.delim(
"data/Ses2_genes.tsv",
stringsAsFactors = FALSE,
na.strings = c("NA", "missing", "Missing")
)
gene_names <- read.delim(
"data/Ses2_geneNames.tsv",
stringsAsFactors = FALSE
)What we want to do now, is to merge the two data frames into one data frame with 6 columns, containing the information from both data frames. We also want to make sure that when R merges the data frames, the correct information is added to the correct row. You will notice that both the genes and gene_names data frames have a column giving the EntrezIDs. This column can be used as the “index” or “ID” column to make sure the correct information is added to each row. We can do this by telling merge() to match rows in the two data frames using EntrezIDs during the merge.
merge() has the following syntax:
merge(
x = # name of the first dataframe to merge
y = # name of the second dataframe to merge
by.x = # name of the column to match, in the first data frame
by.y = # name of the column to match in the second data frame
)Thus, to merge our two dataframes, using the EntrezID column of each data frame to match rows, we can use:
## EntrezID SYMBOL TXCHROM GeneLength Count
## 1 11480 Acvr2a chr2 5681 979
## 2 11634 Aire chr10 1929 15
## 3 12156 <NA> chr2 2515 932
## 4 12290 Cacna1e chr1 12697 0
## 5 12449 Ccnf chr17 3099 214
## 6 12563 Cdh6 chr15 2632 2
## GENENAME
## 1 activin receptor IIA
## 2 autoimmune regulator (autoimmune polyendocrinopathy candidiasis ectodermal dystrophy)
## 3 bone morphogenetic protein 2
## 4 calcium channel, voltage-dependent, R type, alpha 1E subunit
## 5 cyclin F
## 6 cadherin 6You may have noticed that there are 200 rows in the genes data frame but 290 rows in the gene_names data frame. This means that there are more genes in the gene_names data frame than there are in the genes data frame. This means that there are a few ways to merge the two data frames. We can either keep all rows from both data frames, keep only rows where there is a corresponding “index” value in both dataframes or keep only rows from one of the two data frames.
We can specify which rows to keep using the following additional arguments in merge():
merge(
x = # name of the first dataframe to merge
y = # name of the second dataframe to merge
by.x = # name of the column to match, in the first dataframe
by.y = # name of the column to match in the second dataframe
all.x = # logical. If TRUE, keep all rows from the first dataframe,
# even if does not have a matching row in the second dataframe
all.y = # logical. If TRUE, keep all rows from the second dataframe,
# even if does not have a matching row in the second dataframe
)By default, merge() will only keep rows that have corresponding “index” values in both data frames.
Challenge 2.3
Take the
genesdata frame and filter down to only genes from the first 3 chromosomes.Take the result of question 1 and sort it by chromosome then count. i.e. genes in the result should be grouped by chromosome, then count.
Merge the data frames
genesandgene_namesand keep all rows from both data frames.